Variante de la maladie de Creutzfeldt–Jakob


Cet article est orphelin. Moins de trois articles lui sont liés ().
Vous pouvez aider en ajoutant des liens vers [[Variante de la maladie de Creutzfeldt–Jakob]] dans les articles relatifs au sujet.
Variante de la maladie de Creutzfeldt–Jakob
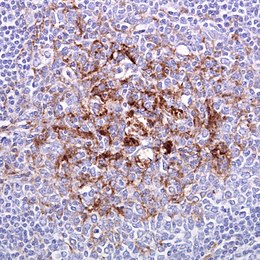
Biopsie de l’amygdale dans la variante de la MCJ. Immunomarquage des protéines prions.
| Causes | Prion |
|---|---|
| Début habituel | Moins de 30 ans [1] |
| Diagnostic | Biopsie cérébrale[1] |
|---|---|
| Traitement | Soins de soutien[2] |
| Pronostic | ~ 13 mois d’espérance de vie[3] |
| Spécialité | Infectiologie |
| Fréquence | Moins de 250 cas signalés en 2012[4] |
|---|
| CIM-10 | A81.0 et F02.1 |
|---|---|
| CIM-9 | 046.1 |
 Mise en garde médicale
Mise en garde médicale
modifier - modifier le code - voir Wikidata (aide)
La variante de la maladie de Creutzfeldt–Jakob est un type de maladie cérébrale appartenant à la famille des encéphalopathies spongiformes transmissibles[4].
Symptômes
Les symptômes comprennent des troubles psychiatriques, des changements de comportement et des sensations douloureuses. Le délai entre l'exposition et l'apparition des symptômes n'est pas clair, mais on estime qu'il est de plusieurs années. L'espérance de vie moyenne après l'apparition des symptômes est de 13 mois[1],[3].
Étiologie
La maladie est causée par des prions, qui sont des protéines mal repliées. La propagation serait principalement due à la consommation de viande de bœuf infectée par l'encéphalopathie spongiforme bovine[4],[5]. On pense également que l'infection nécessite une susceptibilité génétique spécifique[6],[4]. La propagation peut également se produire via des produits sanguins ou du matériel chirurgical contaminé[7].
Diagnostic
Le diagnostic repose sur la biopsie cérébrale mais peut être suspecté sur certains autres critères[1]. Elle est différente de la maladie de Creutzfeldt-Jakob classique, bien que les deux soient dues à des prions[5].
Traitement
Le traitement de la vMCJ comprend des soins de soutien[2].
Fréquence
En 2012, environ 170 cas de vMCJ ont été enregistrés au Royaume-Uni et 50 cas dans le reste du monde. La maladie est devenue moins fréquente depuis 2000. L'âge typique d'apparition est inférieur à 30 ans. Elle a été identifié pour la première fois en 1996 par l'Unité nationale de surveillance de la MCJ à Édimbourg, en Écosse[4],[1].
Références
- ↑ a b c d et e (en-US) « Classic CJD versus Variant CJD » [archive du ], sur CDC, (consulté le )
- ↑ a et b (en-US) « Treatment Variant Creutzfeldt-Jakob Disease » [archive du ], sur CDC, (consulté le )
- ↑ a et b (en-US) « Clinical and Pathologic Characteristics | Variant Creutzfeldt-Jakob Disease, Classic (CJD) » [archive du ], sur CDC, (consulté le )
- ↑ a b c d et e (en) JW Ironside, « Variant Creutzfeldt–Jakob disease: an update. », Folia Neuropathologica, vol. 50, no 1, , p. 50–6. (PMID 22505363)
- ↑ a et b (en-US) « About vCJD » [archive du ], CDC, (consulté le )
- ↑ (en) Ironside, « Variant Creutzfeldt–Jakob disease », Haemophilia, vol. 16 Suppl 5, , p. 175–80. (PMID 20590878, DOI 10.1111/j.1365-2516.2010.02317.x)
- ↑ (en) Fred F. Ferri, Ferri's Clinical Advisor 2018 E-Book: 5 Books in 1, Elsevier Health Sciences, (ISBN 9780323529570, lire en ligne [archive du ]), p. 343
Liens externes
- Ressources relatives à la santé
 :
: - Genetic and Rare Diseases Information Center
- ICD-10 Version:2016
- ICD9Data.com
- Medical Dictionary for Regulatory Activities Terminology
- Notice dans un dictionnaire ou une encyclopédie généraliste :
- Britannica
 Portail de la médecine
Portail de la médecine  Portail des neurosciences
Portail des neurosciences











